














Data science and analytics
An evolving experiment: lifecycle of a data science project

Nearly a hundred years ago, antibiotics became mainstream. Since then, antibiotics have saved countless lives. However, its widespread and irrational usage over the last century has made bacteria resistant to them. It is estimated this could be a cause for millions of deaths by 2050.
In this context, antimicrobial peptides (AMP) are emerging as potential alternatives to antibiotics. However, their underlying complex patterns makes understanding them time-consuming.
This is where machine learning (ML) algorithms can help:
Techniques like alphabet reduction can be applied to group amino acids based on their physicochemical properties
Distributed vector representation can then be used to transform peptides into numeric forms for ML algorithms
Let us take a closer look at the two methodologies for their value in classifying antimicrobial peptides.
Amino acids in peptides are responsible for their structure and function. Every amino acid has unique characteristics generally determined by its side chains.

Fig.1 Reduced peptide sequence splitting: Based on the hydropathy index of each of the amino acids they are grouped into A, B, C and D groups. “Original sequence” refers to an antimicrobial peptide which is then reduced to ABCD based on the hydropathy grouping forming the reduced sequence.
An important outcome of applying this technique is its ability to identify properties that will abstract the required information. In this case, the properties chosen were hydropathy index, conformational similarity, contact energy and substitution matrix. The alphabet reduction technique was applied to extract features based on these properties and then convert them to a numerical form using distributed vector representation.
Just like letters of a language string together to form a word, amino acids form peptides and proteins. Using vector representation which is commonly used in natural language processing helps identify unique patterns in peptide sequences more effectively. Let us take a look at how it works.
Machine learning algorithms need data in numerical format. As a first step, non-numerical data such as text or sequences, need to be converted into a numerical form, or embeddings. An NLP technique — Word2vec — can create word embeddings, the numerical representation of a word, also known as vectors.
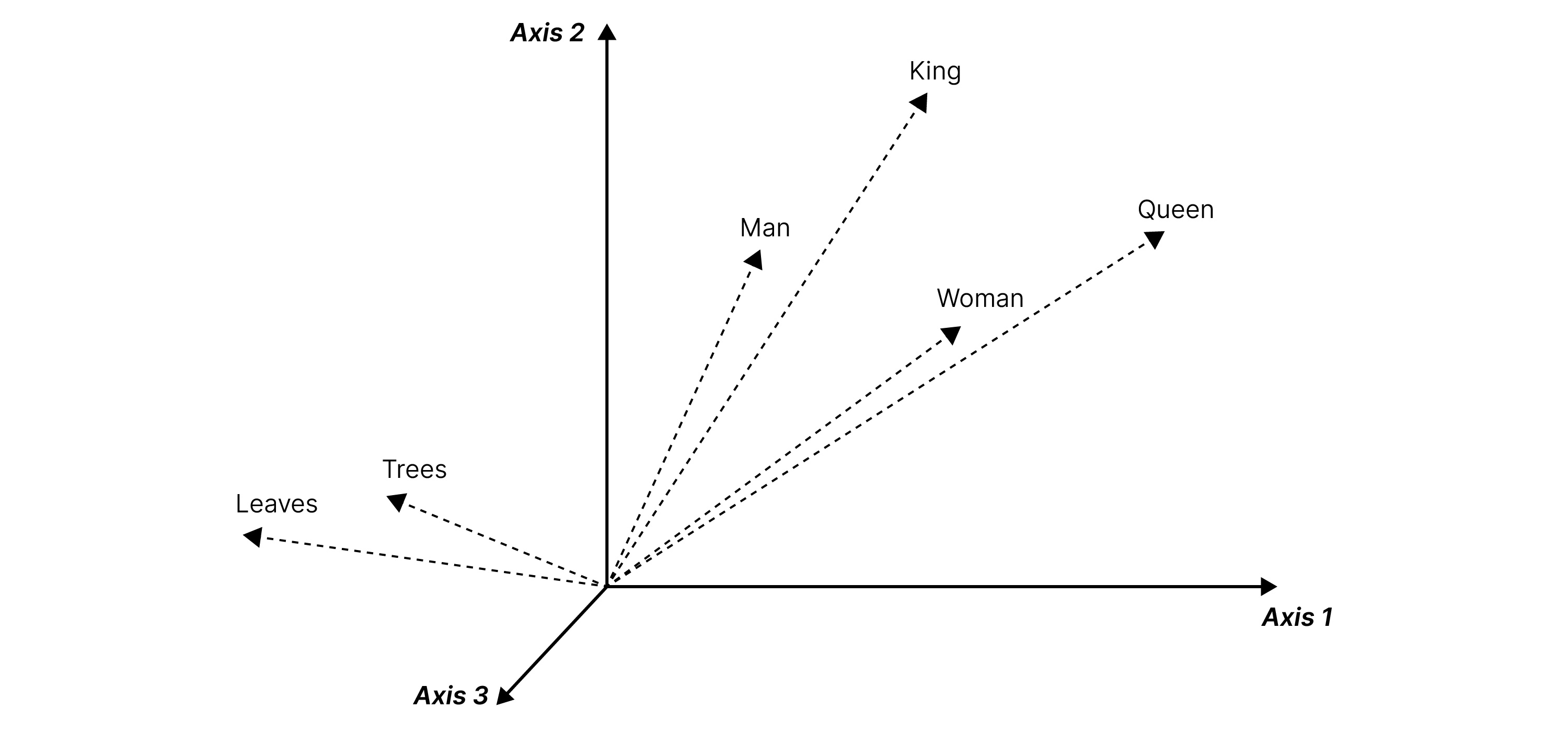
Fig. 2 Word vectors
For example, in the above figure, we see the words ‘king’ and ‘queen’ have their values close to one another in the vector space. ‘Man’ is closer to ‘king’ than ‘queen’. All these are very far from ‘leaves’, whose vector representation is closer to ‘trees’. In these embeddings, the meaning of a word is characterized by its context, or its neighboring words. This can also be applied to the study of peptides.
Each peptide sequence is embedded in an n-dimensional vector. Unique patterns in peptide sequences are sought to facilitate interpretations based on the properties of every amino acid (i.e., the context). ProtVec is used to create these representations to understand the biophysical and biochemical patterns in each of the sequences.
Similar peptides tend to have similar vector representation. Thus, peptides that show antimicrobial properties have similar biophysical and biochemical patterns represented by similar vectors compared to non-antimicrobial peptides. This makes it easier for the machine learning algorithm to classify these peptides as they lie closer to each other in the vector space.
A reduced vector representation can be created by applying the alphabet reduction technique and then converting them to a numerical form using distributed vector representation. This can be done for each of the physicochemical properties and their multiple combinations. For example, hydropathy + conformation similarity is one combination, hydropathy + contact energy is another. All such possibilities led to 256 precise combinations.

Swissport sequences Corpus: 550K sequences from Swissport
↓
Alphabet reduction*
↓
Reduced corpus of Swissport sequences
↓
Word2Vec
↓
High dimensional vector representation of entire swissport
↓
Lookup table
Antimicrobial peptides
↓
Alphabet reduction*
↓
Reduced AMP sequences
↓
Lookup table
↓
High dimension vector representation of antimicrobial peptides
↓
Classification using various ML algorithm
It is possible to perform the binary classification of AMP in two steps by using random forest and deep neural networks.
Vector representation of sequences in the curated dataset using the embeddings created from the Swiss-Prot data, with and without reduction techniques (left)
Machine learning models to classify the sequences using their vector representations (right)
It is possible to create models based on all the 256 combinations to identify the right properties for classifying the antimicrobial peptides. The models can help identify the important properties that contribute most to the classification of AMPs.
The combination of substitution matrix, ProtVec and contact energy gave us the best results. With this, we interpret that the information about evolutionary conserved residues (coming from substitution matrix), protvec (distributed vector representation) and energy between neighboring amino acids (coming from the contact energy) helps in identifying the peptides as AMP. The final model has an accuracy of 97.94% with a Matthews Correlation Coefficient (MCC) of 0.94.
Scientists and pharma researchers are welcome to use this model to identify antimicrobial activity for their sequences. The confirmed sequences can be further studied for their potential as drug candidates against microbes. Such computational methods are of immense use in the long and complex process of drug discovery where they can not only reduce the time taken in laboratory activities but also identify diverse data driven patterns and features otherwise overlooked.
This article is an adaptation of a research paper by Shraddha Surana, Digvijay Gunjal, Divye Singh, Pooja Arora, and Jayaraman Valadi, published in the IEEE International Conference on Bioinformatics and Biomedicine (BIBM) - 2020. You can read the entire research paper, here.
Disclaimer: The statements and opinions expressed in this article are those of the author(s) and do not necessarily reflect the positions of Thoughtworks.

